
A manifold fitting approach for single-cell RNA sequencing data analysis
September 06, 2024Researchers at the National University of Singapore (NUS) have developed a technique that accurately characterises single-cell RNA sequencing (scRNA-seq) data using manifold fitting. This innovation promises to speed up and improve the accuracy of data analysis, aiding biomedical research in areas like cancer and Alzheimer’s disease.
sc-RNA sequencing has become a crucial tool in genomic research, offering unprecedented insights into cellular diversity and disease mechanisms. However, the inherent noise in scRNA-seq data due to biological variability and technical errors, presents a significant challenge arising from its high-dimensional nature and inherent complexity. Conventional analytical approaches – ranging from genomic imputation and graph-based techniques to deep learning algorithms – often fail to decipher the intricate patterns embedded within this data. In response to these limitations, researchers have tried using manifold-learning strategies to overcome these issues, but these methods also have limitations, often losing important information or producing unclear results.
The team, led by Associate Professor Zhigang YAO from the Department of Statistics and Data Science at NUS, with his research fellow Bingjie LI and PhD student Yukun LU, pioneered a new method called scAMF (Single-cell Analysis via Manifold Fitting) to analyse scRNA-seq data more effectively (see Figure for illustration). scAMF works by fitting a low-dimensional manifold within the high-dimensional space where the gene expression data is measured. By doing so, scAMF effectively reduces noise while preserving crucial biological information. This allows for more accurate characterisation of cell types and states.
The key innovation of scAMF lies in its ability to improve the spatial distribution of the data, bringing gene expression vectors of cells from the same type closer together while maintaining clear separation between different cell types. The method employs a unique combination of data transformation, manifold fitting using shared nearest neighbour metrics, and unsupervised clustering validation. Notably, scAMF consistently outperforms existing single-cell analysis methods, including deep learning approaches, across a wide range of datasets in terms of clustering efficiency and data visualisation clarity. This research work is in collaboration with Professor Shing-Tung YAU from Tsinghua University.
Their findings have been published in the Proceedings of the National Academy of Sciences of the United States of America.
Prof Yao highlighted the significance of their work, and said, “By accurately fitting manifolds to scRNA-seq data, we can reduce data dimensionality while preserving crucial information, including the underlying gene expression patterns. This represents a major leap in scRNA-seq analysis, enhancing both accuracy and efficiency.”
“By providing a solution that overcomes the limitations of previous methods, our research paves the way for enhanced single-cell analysis and offers valuable insights for diverse applications in genomics and beyond,” added Prof Yao.
The research team is actively exploring new applications of their framework to tackle even more complex biological datasets. A key focus is the development of a multi-resolution cell analysis framework based on scAMF. This advanced framework aims to identify rare cell populations and contribute to the construction of comprehensive cell atlases. The multi-resolution approach will allow researchers to analyse cellular heterogeneity at various levels of granularity, from broad cell types to subtle subpopulations. This is particularly crucial for identifying rare cell types, such as stem cells or certain immune cell subtypes, which play critical roles in biological processes and disease mechanisms despite being low in abundance.
Prof Yao is scheduled to deliver an invited lecture on this work at the upcoming Second Conference on Geometry and Statistics in China, to be hosted in Shanghai this winter.
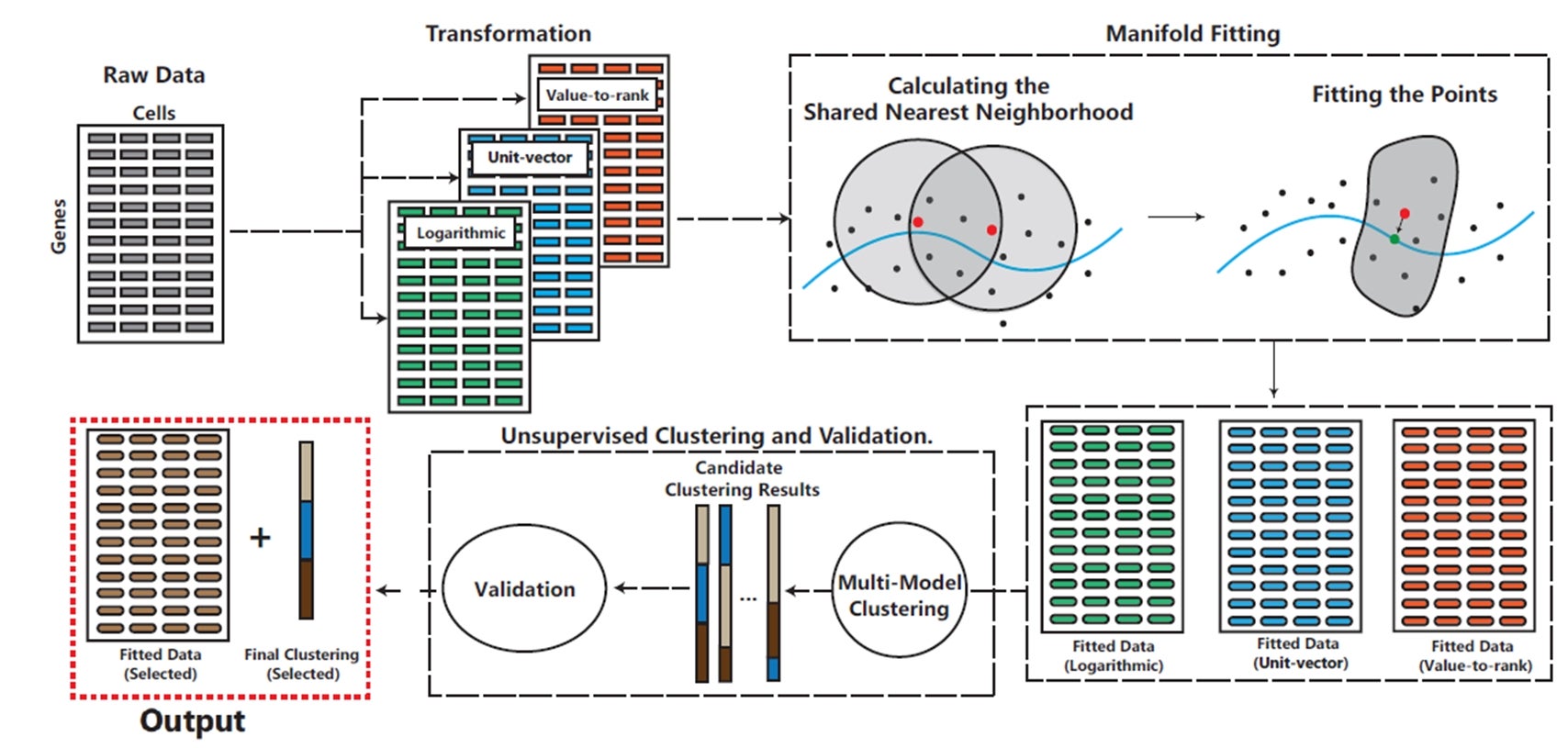
Figure shows a schematic overview of the scAMF pipeline. The process begins with data transformation using three methods: value-to-rank, unit-vector, and logarithmic. This is followed by manifold fitting to denoise the data while preserving its structure. The fitted data then undergoes multiple clustering methods, with an unsupervised validation index selecting the optimal clustering result. The final outputs are the fitted data and cell type assignments. [Credit: Proceedings of the National Academy of Sciences of the United States of America]
Reference
Yao Z*; Li B; Lu Y; Yau ST*, “Single-Cell Analysis via Manifold Fitting: A Framework for RNA Clustering and Beyond”, Proceedings of the National Academy of Sciences of the United States of America DOI: 10.1073/pnas.2400002121 Published: 2024.
