
Directing group-free alkene dicarbofunctionalization through catalyst control
December 24, 2021NUS chemists have developed a new “catalyst control” blueprint to overcome a longstanding challenge in the site-selective dicarbofunctionalization of unactivated alkenes, by enabling the reaction to proceed in the absence of directing or activating groups to access valuable sp3-hybridised carbon frameworks.
Multicomponent catalytic processes that can form multiple carbon–carbon bonds in a single step under mild conditions are highly sought-after for generating molecular complexity in chemical synthesis. One such class of reactions involves the transition-metal-catalysed incorporation of carbogenic units across readily available alkene feedstocks. A key challenge is the ability to control the regioselectivity of olefin addition, especially in situations when the two newly introduced motifs possess similar electronic and/or steric attributes. This problem is further exacerbated in reactions involving the less reactive (unactivated) aliphatic alkenes, which suffer from poor regiochemical control. Conventionally, “substrate control” and “auxiliary control” strategies were employed to implement efficient and site-selective dicarbofunctionalizations across aliphatic olefins (see Figure 1). Unfortunately, both approaches require contrived substrate modifications which are costlier, generate excessive waste and limit practicality.
A research team led by Prof KOH Ming Joo, from the Department of Chemistry, National University of Singapore, has developed a catalytic regime that completely eliminates the need for pre-installed functionalities or directing auxiliaries (see Figure 2). This was achieved by leveraging a dimeric nickel(I) catalyst bearing a sterically encumbered N-heterocyclic carbene (NHC) ligand, which would first react with an aryl triflate and undergo branched-selective carbonickelation via transition state TS A across the alkene, so as to minimise steric repulsions that exist between the sizeable ligand and the alkene substituent(s) in the disfavoured transition state TS A’. Subsequent transmetallation with a Grignard or zinc reagent and reductive elimination furnishes the desired adduct in high efficiency and site selectivity. The regiochemical outcome of these transformations is further supported by density functional theory (DFT) calculations performed by Prof Osvaldo GUTIERREZ, a long-time collaborator from the University of Maryland and currently at Texas A&M University.
Prof Koh said, “With the new Ni-catalysed method, it is not necessary to pre-introduce an activating or directing unit on the alkene substrate to enhance reaction efficiency or control the regioselectivity of dicarbofunctionalization. This provides significant merits in terms of cost savings, step economy and waste reduction.”
“Our catalytic system can potentially accelerate the assembly of drug-like building blocks for compound library screening in drug discovery programmes. We expect the “catalyst control” concept that we conceived in our study to pave the way for the future development of general alkene addition processes that no longer rely on activating units within the substrate for high efficiency and selectivity,” added Prof Koh.
The research team is currently expanding the scope of this work towards the synthesis of enantioenriched compounds.
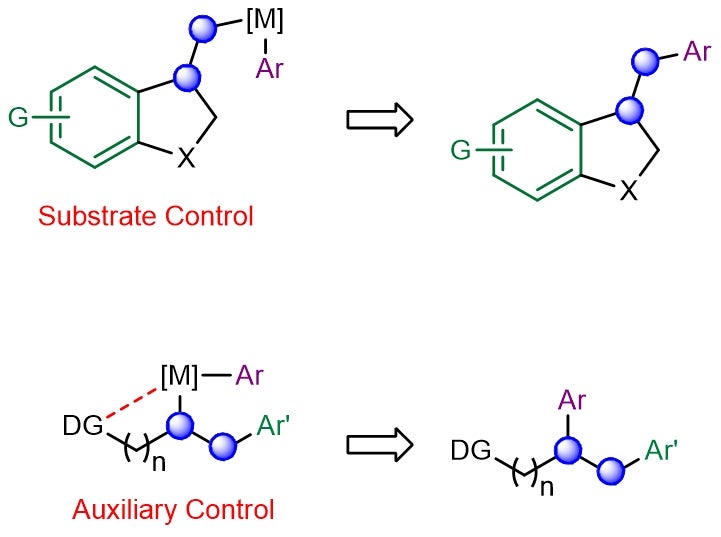
Figure 1: Schematic showing the existing substrate- and auxiliary-controlled strategies that induce high regioselectivity in transition metal-catalysed alkene dicarbofunctionalization. [Credit: Nature Chemistry]
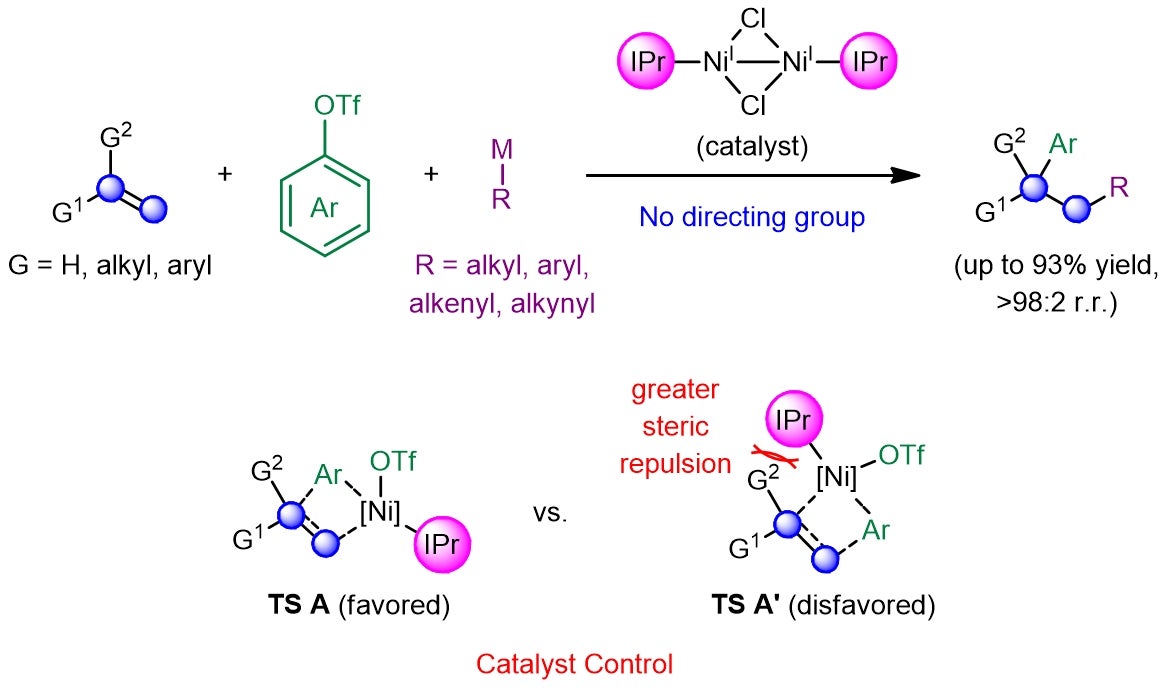 Figure 2: Schematic showing the development of a catalyst-controlled regime that promotes efficient and site-selective dicarbofunctionalization of alkenes in the absence of any activating or directing groups. [Credit: Nature Chemistry]
Figure 2: Schematic showing the development of a catalyst-controlled regime that promotes efficient and site-selective dicarbofunctionalization of alkenes in the absence of any activating or directing groups. [Credit: Nature Chemistry]
Reference
Wang H; Liu C; Martin RT; Gutierrez O*; Koh MJ*, “Directing-group-free catalytic dicarbofunctionalization of unactivated alkenes” NATURE CHEMISTRY DOI: 10.1038/s41557-021-00836-6 Published: 2021.
This research achievement is also highlighted by Chemical and Engineering News (C&EN).
